Research published today in Respiratory Research presents findings from the IPF-PRO Registry, a nationwide US registry collecting data on the clinical course of Idiopathic pulmonary fibrosis (IPF) and its impact on patients.

Courtesy of Dr. Laurie Snyder
Idiopathic pulmonary fibrosis, or IPF, is a rare, debilitating lung disease that mainly affects individuals over the age of 60. IPF is characterized by progressive fibrosis (scarring) of the lungs, which leads to a loss of lung volume and an impaired ability of the lungs to exchange gases. Ultimately IPF is fatal.
While IPF is always progressive, the rate at which it progresses differs between patients. At present, it is not possible to predict the course of the disease for an individual patient. This presents challenges both for the patients living with the disease and the healthcare professionals who care for them.
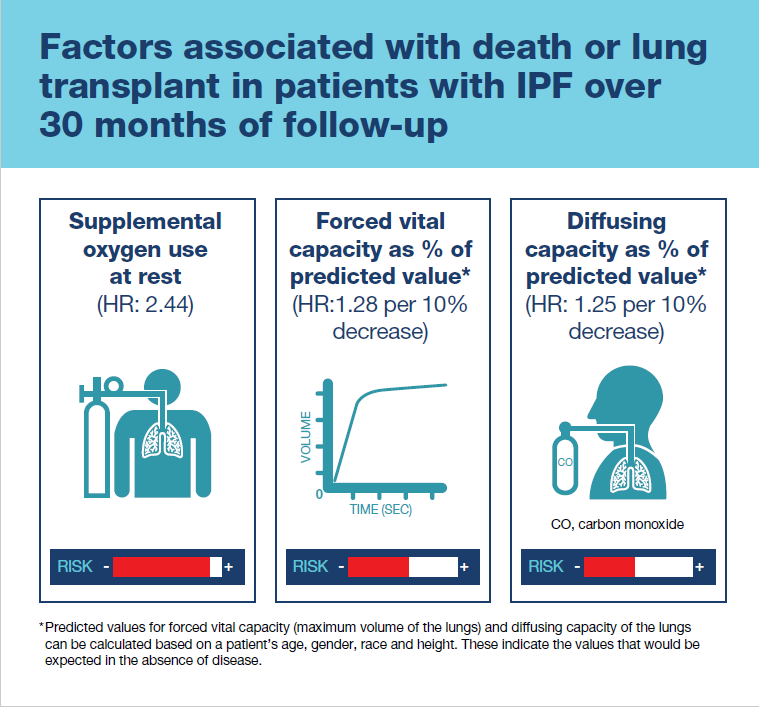
The strongest risk factor for death or lung transplant was use of supplemental oxygen at rest.
Patient registries are an important source of information on rare diseases like IPF. The IPF-PRO Registry is a nationwide US registry that was set up to collect data on the clinical course of IPF and its impact on patients. It is co-ordinated by the Duke Clinical Research Institute in North Carolina and funded by Boehringer Ingelheim Pharmaceuticals, Inc.
To date, the IPF-PRO Registry has enrolled over 1000 patients with IPF that was diagnosed or confirmed at the enrolling center within the past 6 months. These patients are now being followed prospectively, with data collected on their lung function, symptoms, quality of life and treatment.
In our recent paper, we present data from the first 662 patients enrolled in the IPF-PRO Registry. We looked at which of the characteristics of these patients at enrollment into the registry were associated with mortality or lung transplant over a follow-up period of 30 months. We included lung transplant as well as mortality as an outcome, as lung transplant is a marker of disease progression that would otherwise be expected to result in death.
We found that the patients in our study had about a 50% probability of dying or having a lung transplant over 30 months. The strongest risk factor for death or lung transplant was use of supplemental oxygen at rest. We also showed that patients with worse lung function, based on a lower forced vital capacity (maximum volume of the lungs) or lower capacity of the lungs to exchange oxygen (diffusing capacity) were at greater risk of death or lung transplant.
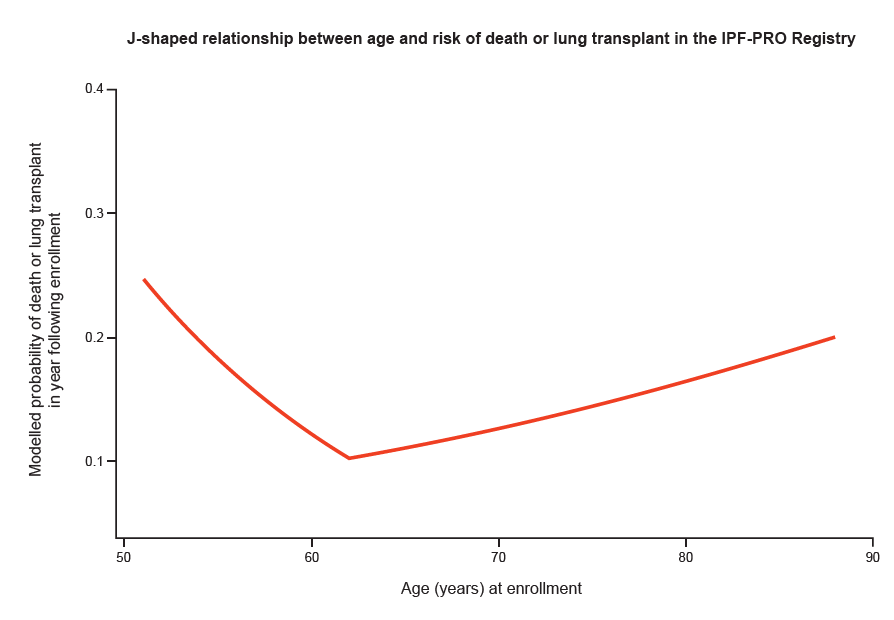
When we looked at the relationship between age and the risk of death or lung transplant, we found a J-shaped relationship. Patients who were aged around 60 had the lowest risk, while patients who were older had an increased risk and the youngest patients had the highest risk of all. More research is needed, but possibly the youngest patients with IPF had a familial (genetic) form of the disease, which progresses more quickly.
We’ve also been studying whether worse symptoms or quality of life in patients in the registry are predictors of death or lung transplant and we plan to publish those data soon. Moving forward, we are going to be using data from the IPF-PRO Registry to look at whether changes in clinical or patient-reported factors are better predictors of mortality than values at a single point in time. These data will help give us a better understanding of the course of this devastating disease.